New year, new challenge: mass spectra.
To calculate the theoretical isotope pattern for a given molecule in PostgreSQL you need two things:
- a cheminformatics toolkit to determine the element composition of the molecule and it's exact mass
- a usably fast and accurate algorithm to infer the isotope pattern from the composition
Then, the two have to be made working together. Like so:
typedef struct
{
size_t num_entries;
double *mz;
double *intensity;
double *intensity_normalized;
unsigned int *md;
} _ISOTOPE_PATTERN;
{
size_t num_entries;
double *mz;
double *intensity;
double *intensity_normalized;
unsigned int *md;
} _ISOTOPE_PATTERN;
extern "C" _ISOTOPE_PATTERN *
ob_molfile_to_isotope_pattern(char *molfile, int charge, double normal)
{
_ISOTOPE_PATTERN *retval;
vector msa_mz;
vector msa_abundance;
vector composition;
vector m;
const double limit = 10e-30;
OBMol mol;
OBConversion conv;
string tmpStr (molfile);
istringstream molstream (tmpStr);
double mass_diff, monoisotopic_mass, scale = 1.0, delta = numeric_limits::max();
int i=0;
//Initialize composition vector to all zeroes
for(unsigned int i=0; i composition.push_back(0);
conv.SetInAndOutFormats("MDL",NULL);
conv.Read(&mol, &molstream);
if (mol.Empty ())
return NULL;
// Add missing hydrogens, if any
mol.AddHydrogens(false, false); // All Hs, no PH correction
//Iterate over atoms and update the composition vector accordingly
FOR_ATOMS_OF_MOL(atom, mol)
{
switch (atom->GetAtomicNum())
{
case 1: // H
composition[0]++;
break;
case 6: // C
composition[1]++;
break;
case 7: // N
composition[2]++;
break;
case 8: // O
composition[3]++;
break;
case 16: // S
composition[4]++;
break;
case 35: // Br
composition[5]++;
break;
case 17: // Cl
composition[6]++;
break;
case 53: // I
composition[7]++;
break;
case 15: // P
composition[8]++;
break;
case 9: // F
composition[9]++;
break;
default: // Others ignored
break;
}
}
// Calculate isotope patterns with MERCURY7
if(0 != mercury::mercury(msa_mz, msa_abundance, composition, charge, limit))
{
return NULL;
}
monoisotopic_mass = mol.GetExactMass();
// Allocate return value and copy values
retval = (_ISOTOPE_PATTERN*) calloc(1,sizeof(_ISOTOPE_PATTERN));
retval->num_entries = msa_mz.size();
retval->mz=(double*) calloc(msa_mz.size(), sizeof(double));
retval->intensity=(double*) calloc(msa_abundance.size(), sizeof(double));
retval->intensity_normalized=(double*) calloc(msa_abundance.size(), sizeof(double));
retval->md=(unsigned int*) calloc(msa_abundance.size(), sizeof(unsigned int));
copy(msa_mz.begin(), msa_mz.end(), retval->mz);
copy(msa_abundance.begin(), msa_abundance.end(), retval->intensity);
copy(msa_abundance.begin(), msa_abundance.end(), retval->intensity_normalized);
for(std::vector::iterator it = msa_mz.begin(); it != msa_mz.end(); it++)
{
mass_diff = *it-monoisotopic_mass;
m.push_back(mass_diff);
if(abs(mass_diff) < delta)
{
delta = abs(mass_diff);
scale = msa_abundance[i];
}
i++;
}
scale = normal / scale;
for(i=0; retval->num_entries; i++)
{
retval->intensity_normalized[i] *= scale;
}
copy(m.begin(), m.end(), retval->md);
return retval;
}
ob_molfile_to_isotope_pattern(char *molfile, int charge, double normal)
{
_ISOTOPE_PATTERN *retval;
vector msa_mz;
vector msa_abundance;
vector composition;
vector m;
const double limit = 10e-30;
OBMol mol;
OBConversion conv;
string tmpStr (molfile);
istringstream molstream (tmpStr);
double mass_diff, monoisotopic_mass, scale = 1.0, delta = numeric_limits::max();
int i=0;
//Initialize composition vector to all zeroes
for(unsigned int i=0; i composition.push_back(0);
conv.SetInAndOutFormats("MDL",NULL);
conv.Read(&mol, &molstream);
if (mol.Empty ())
return NULL;
// Add missing hydrogens, if any
mol.AddHydrogens(false, false); // All Hs, no PH correction
//Iterate over atoms and update the composition vector accordingly
FOR_ATOMS_OF_MOL(atom, mol)
{
switch (atom->GetAtomicNum())
{
case 1: // H
composition[0]++;
break;
case 6: // C
composition[1]++;
break;
case 7: // N
composition[2]++;
break;
case 8: // O
composition[3]++;
break;
case 16: // S
composition[4]++;
break;
case 35: // Br
composition[5]++;
break;
case 17: // Cl
composition[6]++;
break;
case 53: // I
composition[7]++;
break;
case 15: // P
composition[8]++;
break;
case 9: // F
composition[9]++;
break;
default: // Others ignored
break;
}
}
// Calculate isotope patterns with MERCURY7
if(0 != mercury::mercury(msa_mz, msa_abundance, composition, charge, limit))
{
return NULL;
}
monoisotopic_mass = mol.GetExactMass();
// Allocate return value and copy values
retval = (_ISOTOPE_PATTERN*) calloc(1,sizeof(_ISOTOPE_PATTERN));
retval->num_entries = msa_mz.size();
retval->mz=(double*) calloc(msa_mz.size(), sizeof(double));
retval->intensity=(double*) calloc(msa_abundance.size(), sizeof(double));
retval->intensity_normalized=(double*) calloc(msa_abundance.size(), sizeof(double));
retval->md=(unsigned int*) calloc(msa_abundance.size(), sizeof(unsigned int));
copy(msa_mz.begin(), msa_mz.end(), retval->mz);
copy(msa_abundance.begin(), msa_abundance.end(), retval->intensity);
copy(msa_abundance.begin(), msa_abundance.end(), retval->intensity_normalized);
for(std::vector::iterator it = msa_mz.begin(); it != msa_mz.end(); it++)
{
mass_diff = *it-monoisotopic_mass;
m.push_back(mass_diff);
if(abs(mass_diff) < delta)
{
delta = abs(mass_diff);
scale = msa_abundance[i];
}
i++;
}
scale = normal / scale;
for(i=0; retval->num_entries; i++)
{
retval->intensity_normalized[i] *= scale;
}
copy(m.begin(), m.end(), retval->md);
return retval;
}
This parses a V2000 or V3000 molfile into a molecule object, iterates over the atoms and generates the elemental composition for Mercury7, calculates the exact mass and the theoretical isotope pattern. It then stores the m/z vector, the peak vector and the derived peak vector normalized to some value, e.g. 100%, and the mass distance relative to the base peak m/z value. All of this is stored in the C structure _ISOTOPE_PATTERN in order to transfer it from C++ to the C code of PostgreSQL.
Over to the PostgreSQL side of things :-)
PG_FUNCTION_INFO_V1 (pgchem_isotope_pattern);
Datum
pgchem_isotope_pattern (PG_FUNCTION_ARGS)
{
_ISOTOPE_PATTERN *isotope_pattern = NULL;
_ISOTOPE_PATTERN *pattern_copy = NULL;
FuncCallContext *ctx=NULL;
MemoryContext original_ctx=NULL;
DECOMPRESSED_DATA *original_data=NULL;
char *tmpMolfile=NULL;
if(SRF_IS_FIRSTCALL())
{
MOLECULE *arg_molecule = PG_GETARG_MOLECULE_P (0);
int32 arg_charge = PG_GETARG_INT32 (1);
float8 arg_normal = PG_GETARG_FLOAT8 (2);
ctx = SRF_FIRSTCALL_INIT();
original_ctx = MemoryContextSwitchTo(ctx->multi_call_memory_ctx);
if(arg_molecule->original_format==FORMAT_V2000 || arg_molecule->original_format==FORMAT_V3000)
{
original_data = decompress_data(CIPTR(arg_molecule),arg_molecule->compressed_sizeo, arg_molecule->sizeo);
tmpMolfile = strdup(original_data->decompressed_data);
}
else
{
tmpMolfile = ob_smiles_to_V2000 (SMIPTR(arg_molecule));
}
isotope_pattern = ob_molfile_to_isotope_pattern(tmpMolfile, arg_charge, arg_normal);
if(NULL!=tmpMolfile)
{
free(tmpMolfile);
}
if(NULL == isotope_pattern)
{
elog (ERROR, "Could not generate isotope pattern!");
PG_RETURN_NULL();
}
pattern_copy = (_ISOTOPE_PATTERN*) palloc0(sizeof(_ISOTOPE_PATTERN));
pattern_copy->mz = (double*) palloc0(isotope_pattern->num_entries*sizeof(double));
pattern_copy->intensity = (double*) palloc0(isotope_pattern->num_entries*sizeof(double));
pattern_copy->intensity_normalized = (double*) palloc0(isotope_pattern->num_entries*sizeof(double));
pattern_copy->md = (unsigned int*) palloc0(isotope_pattern->num_entries*sizeof(unsigned int));
pattern_copy->num_entries = isotope_pattern->num_entries;
memcpy(pattern_copy->mz, isotope_pattern->mz, isotope_pattern->num_entries*sizeof(double));
memcpy(pattern_copy->intensity, isotope_pattern->intensity, isotope_pattern->num_entries*sizeof(double));
memcpy(pattern_copy->intensity_normalized, isotope_pattern->intensity_normalized, isotope_pattern->num_entries*sizeof(double));
memcpy(pattern_copy->md, isotope_pattern->md, isotope_pattern->num_entries*sizeof(unsigned int));
if(NULL != isotope_pattern->md)
free(isotope_pattern->md);
if(NULL != isotope_pattern->mz)
free(isotope_pattern->mz);
if(NULL != isotope_pattern->intensity)
free(isotope_pattern->intensity);
if(NULL != isotope_pattern->intensity_normalized)
free(isotope_pattern->intensity_normalized);
if(NULL != isotope_pattern)
free(isotope_pattern);
isotope_pattern = NULL;
ctx->max_calls = pattern_copy->num_entries;
ctx->user_fctx = pattern_copy;
if(get_call_result_type(fcinfo, NULL, &ctx->tuple_desc) != TYPEFUNC_COMPOSITE)
{
elog (ERROR, "Calling the isotope pattern SRF in a context that does not expect tuples in return is not supported!");
}
BlessTupleDesc(ctx->tuple_desc);
MemoryContextSwitchTo(original_ctx);
}
ctx = SRF_PERCALL_SETUP();
isotope_pattern = ctx->user_fctx;
if(ctx->call_cntr < ctx->max_calls)
{
HeapTuple rettuple = NULL;
Datum *retvals;
bool *retnulls;
retvals = (Datum*) palloc0(4*sizeof(Datum));
retnulls = (bool*) palloc0(4*sizeof(bool));
retvals[0] = Float8GetDatum(isotope_pattern->mz[ctx->call_cntr]);
retvals[1] = Float8GetDatum(isotope_pattern->intensity[ctx->call_cntr]);
retvals[2] = Float8GetDatum(isotope_pattern->intensity_normalized[ctx->call_cntr]);
retvals[3] = UInt8GetDatum(isotope_pattern->md[ctx->call_cntr]);
retnulls[0] = false;
retnulls[1] = false;
retnulls[2] = false;
retnulls[3] = false;
rettuple = heap_form_tuple(ctx->tuple_desc, retvals, retnulls);
pfree(retvals);
pfree(retnulls);
SRF_RETURN_NEXT(ctx, HeapTupleGetDatum(rettuple));
}
else
{
SRF_RETURN_DONE(ctx);
}
}
Datum
pgchem_isotope_pattern (PG_FUNCTION_ARGS)
{
_ISOTOPE_PATTERN *isotope_pattern = NULL;
_ISOTOPE_PATTERN *pattern_copy = NULL;
FuncCallContext *ctx=NULL;
MemoryContext original_ctx=NULL;
DECOMPRESSED_DATA *original_data=NULL;
char *tmpMolfile=NULL;
if(SRF_IS_FIRSTCALL())
{
MOLECULE *arg_molecule = PG_GETARG_MOLECULE_P (0);
int32 arg_charge = PG_GETARG_INT32 (1);
float8 arg_normal = PG_GETARG_FLOAT8 (2);
ctx = SRF_FIRSTCALL_INIT();
original_ctx = MemoryContextSwitchTo(ctx->multi_call_memory_ctx);
if(arg_molecule->original_format==FORMAT_V2000 || arg_molecule->original_format==FORMAT_V3000)
{
original_data = decompress_data(CIPTR(arg_molecule),arg_molecule->compressed_sizeo, arg_molecule->sizeo);
tmpMolfile = strdup(original_data->decompressed_data);
}
else
{
tmpMolfile = ob_smiles_to_V2000 (SMIPTR(arg_molecule));
}
isotope_pattern = ob_molfile_to_isotope_pattern(tmpMolfile, arg_charge, arg_normal);
if(NULL!=tmpMolfile)
{
free(tmpMolfile);
}
if(NULL == isotope_pattern)
{
elog (ERROR, "Could not generate isotope pattern!");
PG_RETURN_NULL();
}
pattern_copy = (_ISOTOPE_PATTERN*) palloc0(sizeof(_ISOTOPE_PATTERN));
pattern_copy->mz = (double*) palloc0(isotope_pattern->num_entries*sizeof(double));
pattern_copy->intensity = (double*) palloc0(isotope_pattern->num_entries*sizeof(double));
pattern_copy->intensity_normalized = (double*) palloc0(isotope_pattern->num_entries*sizeof(double));
pattern_copy->md = (unsigned int*) palloc0(isotope_pattern->num_entries*sizeof(unsigned int));
pattern_copy->num_entries = isotope_pattern->num_entries;
memcpy(pattern_copy->mz, isotope_pattern->mz, isotope_pattern->num_entries*sizeof(double));
memcpy(pattern_copy->intensity, isotope_pattern->intensity, isotope_pattern->num_entries*sizeof(double));
memcpy(pattern_copy->intensity_normalized, isotope_pattern->intensity_normalized, isotope_pattern->num_entries*sizeof(double));
memcpy(pattern_copy->md, isotope_pattern->md, isotope_pattern->num_entries*sizeof(unsigned int));
if(NULL != isotope_pattern->md)
free(isotope_pattern->md);
if(NULL != isotope_pattern->mz)
free(isotope_pattern->mz);
if(NULL != isotope_pattern->intensity)
free(isotope_pattern->intensity);
if(NULL != isotope_pattern->intensity_normalized)
free(isotope_pattern->intensity_normalized);
if(NULL != isotope_pattern)
free(isotope_pattern);
isotope_pattern = NULL;
ctx->max_calls = pattern_copy->num_entries;
ctx->user_fctx = pattern_copy;
if(get_call_result_type(fcinfo, NULL, &ctx->tuple_desc) != TYPEFUNC_COMPOSITE)
{
elog (ERROR, "Calling the isotope pattern SRF in a context that does not expect tuples in return is not supported!");
}
BlessTupleDesc(ctx->tuple_desc);
MemoryContextSwitchTo(original_ctx);
}
ctx = SRF_PERCALL_SETUP();
isotope_pattern = ctx->user_fctx;
if(ctx->call_cntr < ctx->max_calls)
{
HeapTuple rettuple = NULL;
Datum *retvals;
bool *retnulls;
retvals = (Datum*) palloc0(4*sizeof(Datum));
retnulls = (bool*) palloc0(4*sizeof(bool));
retvals[0] = Float8GetDatum(isotope_pattern->mz[ctx->call_cntr]);
retvals[1] = Float8GetDatum(isotope_pattern->intensity[ctx->call_cntr]);
retvals[2] = Float8GetDatum(isotope_pattern->intensity_normalized[ctx->call_cntr]);
retvals[3] = UInt8GetDatum(isotope_pattern->md[ctx->call_cntr]);
retnulls[0] = false;
retnulls[1] = false;
retnulls[2] = false;
retnulls[3] = false;
rettuple = heap_form_tuple(ctx->tuple_desc, retvals, retnulls);
pfree(retvals);
pfree(retnulls);
SRF_RETURN_NEXT(ctx, HeapTupleGetDatum(rettuple));
}
else
{
SRF_RETURN_DONE(ctx);
}
}
This is a set returning function, meaning that it can return multiple rows in one call, i.e. it returns a complete table. Basically it retrieves the molecule argument from PostgreSQL, calls ob_molfile_to_isotope_pattern(), constructs a set of tuples and returns them to PostgreSQL. The basics of set returning functions can be found here and here.
Now we can declare the function to PostgreSQL:
CREATE OR REPLACE FUNCTION isotope_pattern(IN molecule, IN integer DEFAULT 0, IN double precision DEFAULT 100.0)
RETURNS TABLE("m/z" double precision, intensity double precision, intensity_normalized double precision, mdtbp integer) AS
'libpgchem', 'pgchem_isotope_pattern'
LANGUAGE c IMMUTABLE STRICT;
RETURNS TABLE("m/z" double precision, intensity double precision, intensity_normalized double precision, mdtbp integer) AS
'libpgchem', 'pgchem_isotope_pattern'
LANGUAGE c IMMUTABLE STRICT;
and use it:
select * from isotope_pattern('c1ccccc1I'::molecule) where intensity >= 0.01;
| m/z | intensity | intensity_normalized | mdtbp |
| 203.9436 | 0.934689933304048 | 100 | 0 |
| 204.946987674881 | 0.0634781699018538 | 6.79136124612619 | 1 |
Visualized it looks like this:
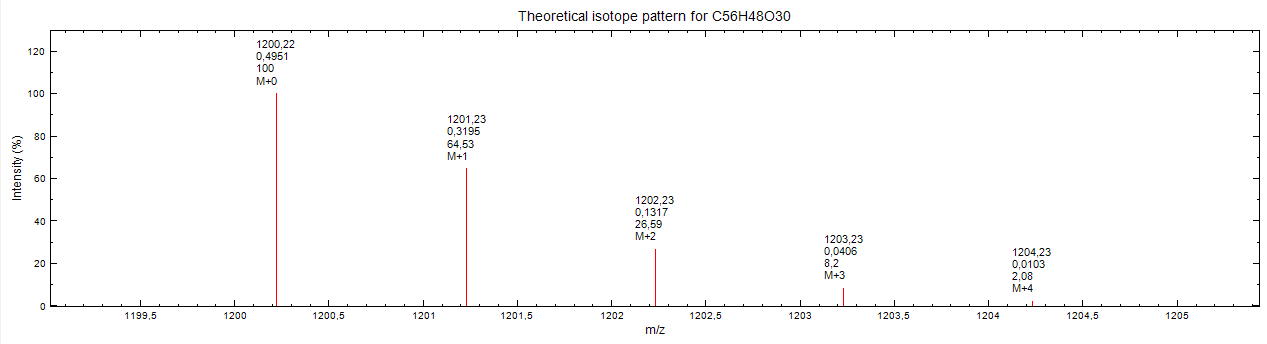
Nice. And everything (except the GUI) in the database.